|
|
|
| When Chemists First "Discovered" NMR |
| |
Abstract
This review charts the early history of NMR from the time that it first
began to be of interest in chemistry. It begins with the efforts of a
few far-sighted chemists to build their own spectrometers, exemplified
by one such laboratory in Oxford in the early 1950s. The key
breakthrough into high-resolution NMR by scientists at Stanford is
highlighted by citing the classic papers written by Jim Arnold and Wes
Anderson, based on their doctoral research. The evolution of
electromagnet and permanent magnet technology is described, culminating
in the introduction of commercial NMR spectrometers which opened the
field to a broad community of organic chemists. The quest for higher
sensitivity led to the introduction of the time averaging technique and
the concept of multichannel excitation, triggering the Fourier transform
revolution and the advent of practical carbon-13 spectroscopy.
|
| |
1. Introduction
When the phenomenon of nuclear magnetic resonance1,2
was first discovered, it was exclusively the province of physicists.
Almost all the known elements possess one or more isotopes with magnetic
properties, and there was a concerted effort to search for these
unknown resonance responses, as there was no way to predict the
frequency at which they would appear in a given magnetic field. Once the
nuclear resonance frequency of a particular nuclear species has been
measured, it defines the magnetic moment. Although frequencies
can be measured with very high precision - magnetic field strengths are
best derived by way of magnetic resonance; consequently the new magnetic
moments were usually related to that of the proton. At the time, the
magnetic moment was believed to represent an important fundamental
property of the nucleus.
This new property aroused intense interest among physicists because it
seemed to offer new insight into nuclear structure. Then (horror of horrors)
evidence began to emerge that these fundamental "constants" were not
really constant at all, but appeared to depend on the nature of the
sample used for the measurement. This disturbing observation is vividly
illustrated by the case of ammonium nitrate, investigated by Proctor and
Yu3 in an attempt to derive an accurate value for the magnetic moment of 14N.
To their consternation they observed two resonance lines. Owing to an
understandable reluctance to accept that this might be a mere chemical
effect, every effort was made to seek a less disturbing explanation. To
this end they placed an order for a sample of 15N-enriched ammonium nitrate in the (vain) hope that the unwanted second resonance line was actually from the 15N isotope. In fact the two resonances represented two different 14N chemical sites in the same molecule. About the same time, Dickinson4 found similar "discrepancies" in the 19F resonance frequencies of several fluorine compounds, Lindstrom5 showed that the ratio of 2H and 1H resonance frequencies differed in water and paraffin oil, and Thomas6 observed further instances where the proton resonance frequencies depended on the sample used. Something was seriously
wrong. How could all the excitement about a fundamental, high-precision
nuclear property of such intrinsic importance be so cruelly dashed by a
mere chemical shift? Most physicists gave up in disgust.
|
| |
2. The Dawn of NMR in Chemistry
Many setbacks in science turn out to have a silver lining. A few
far-sighted scientists began to wonder if this new magnetic resonance
discovery could be applied to chemistry, by concentrating attention on
the chemical effects so despised by the physicists. Rex Richards at
Oxford believed that NMR would be a promising new avenue for chemistry
research, but the physicists he consulted were uniformly discouraging.
Fortunately the world-famous chemist Linus Pauling was visiting Oxford
at the time and he told Rex "Don't believe anything the physicists tell
you", so Rex went ahead. The justification for such optimism was rather
thin at that time, but at least NMR was an open field, and Rex is a
courageous researcher. However in that era, a typical chemistry
laboratory would be mainly devoted to "wet chemistry" and it was really
no place for two-ton magnets. To introduce any large physical machine
into that environment was a real battle, involving banishment to some
dreary basement room that no one else wanted to use. On the positive
side for an NMR pioneer there was a good supply of war-surplus radar
equipment that could be usefully cannibalized, and it was always
possible to ask those now disillusioned physicists for technical advice.
It was the work of George Pake7 that indicated a possible line of attack for chemists. Pake chose to investigate a single crystal of gypsum (CaSO4.2H2O)
where the two protons of the water molecule are the only significantly
magnetic species. He showed that the NMR signal was a doublet of
resonances, representing the fact that each proton in the water molecule
could "see" the two possible orientations (up or down) of its partner.
The splitting of this doublet depended on the orientation of the crystal
with respect to the magnetic field and gave a measure of the
inter-proton distance. An important justification for studying
solid-state NMR was that it filled a serious gap in the coverage of
X-ray crystallography, where the scattering was mainly from the heavier
atoms and it was quite difficult to pin down the location of protons. To
render the method more generally applicable, Pake extended the theory
to powder samples where the spectrum is made up of the superposition of
subspectra from a very large number of microcrystals with all possible
orientations in space. The result was a characteristic broad line shape -
the "Pake doublet". A later collaboration with two chemists, Herbert
Gutowsky and George Kistiakowsky8,
confirmed that this was indeed a valid approach for determining
molecular structure. Chemistry was beginning to gain a foothold in
magnetic resonance.
However it was not a simple task to identify suitable chemical
applications of Pake's work. In Rex Richards' fledgling NMR laboratory
in the 1950's, all the "easy" experiments seemed to have been exhausted.
The remaining chemical samples on offer were either alarmingly toxic or
explosively unstable, and any junior research student took his life
into his own hands. In fact, once an NMR measurement was complete, we
carefully buried our samples (after a brief ceremony) in a grassy slope
adjacent to the laboratory; that lawn still retains some strange
colorations. To make matters worse, the NMR sample had to be cooled to
minimize motional effects, and the only available refrigerants at that
time were liquid oxygen and liquid hydrogen, an unholy alliance. Our
small 12.2 MHz electromagnet was powered by a bank of war-surplus
submarine batteries that had to be recharged overnight (just as in the
original submarines). There was an inevitable slow drift of the magnet
field as the voltage fell off with time. This drift was manually
corrected by a research student monitoring a sensitive mirror
galvanometer, and compensating the downward trend in current by winding a
resistive graphite rod down into a pool of mercury. Just how smoothly
the mercury/carbon interface behaved proved to be critical, and we
discovered that Eagle Turquoise 6H pencil leads were the only reliable
candidates for the graphite rod (the Oxford stationers were bewildered
by this unexplained popularity of one particular pencil). We like to
think of this tedious scheme as a crude fore-runner of the very
successful Varian "superstabilizer" where magnet drift was compensated
electronically, using two photocells to replace the research student,
and a current feedback system.
My own contribution10
to these endeavours was to measure the Pake doublet in a powder sample
of potassium amide (another unstable compound that had to be prepared
and kept under dry nitrogen). Since the spectrometer recorded the first
derivative of the NMR absorption-mode signal, integrations had to be
calculated, using a planimeter that eventually turned out to be faulty
and had to be replaced. In polycrystalline powders the effect of the
dipolar interactions with more-distant protons caused a broadening that
could be represented by a Gaussian function. Consequently the fitting of
the theoretical and experimental lineshapes required some
time-consuming calculations, and the available computers of that era
were essentially hand-cranked adding machines. This expenditure of
blood, sweat and tears was aggravated when a referee (quite reasonably)
demanded that the calculations be repeated with slightly different
combinations of the inter-proton distance and the Gaussian broadening
function in order to obtain an estimate of the accuracy of the result.
After allowance for small torsional and vibration motions of the amide
group, the H-H distance turned out to be 1.63 ± 0.03 Angstrom units, not
exactly a world-shaking discovery. It was this sobering experience that
convinced me that solid-state NMR would always be slow and unrewarding
work. It was not until many years later that innovations by John Waugh11, and Peter Mansfield12
opened up this field enormously, and solid-state NMR became popular
again. Could it be that Linus Pauling's suggestion about NMR and
chemistry had been somewhat over-optimistic?
|
| |
3. The "Do it yourself" era
Initially the most daunting barrier to chemists hoping to try NMR was
the realization that a large magnet and complex electronics would have
to be assembled from scratch; there were no available commercial NMR
spectrometers. Nor was there appreciable funding for purchase of
components, since most chemistry laboratories operated on a shoestring
budget geared to the purchase of glassware and a few chemicals. In the
Physical Chemistry Laboratory at Oxford, Rex Richard did most of the
construction work with his own hands. He acquired an old London taxi,
the kind with a reinforced platform next to the driver to carry large
items of luggage, and used this to transport several large chunks of
soft iron, one at a time, to the local car factory at Cowley for
machining. He wound the energizing coils of the electromagnet by hand,
totalling nine miles of copper wire. All the electronics was from
stripped-down war surplus radar equipment, indeed even our nuts and
bolts had been carefully rescued from these radar sets. (It is
interesting to note that the early pioneers of NMR had all been
participants in wartime radar research, and it was this expertise that
encouraged them to search for magnetic resonance in bulk matter.) It was
clear that the magnet pole caps were critical components as they
determined the eventual uniformity of the field, but the existing
prescriptions for the best geometry and metallurgy somehow smacked of
witchcraft. There are several accounts of endless, patient
hand-polishing of pole faces to achieve a flat surface, and even one
apocryphal story about a field contour map that was misinterpreted, so
that regions making lower contributions to the magnetic field were
ground down even further. The direct current power supply also proved to
be a tricky item, for this is what determines the stability of the
magnetic field. It required a set of large vacuum tubes for current
regulation operating in parallel. There were concerns (later discovered
to be largely unfounded) that switching off the magnet might induce an
unacceptably large surge in current. Eventually our own monster magnet
was installed; basic physics had at last insinuated itself into the
Oxford chemistry laboratory. Some wag attached a notice that read "Magnet, not to be removed from room 16".
This new apparatus was used to investigate "other nuclei" (anything
except the proton) in the liquid state. Surely, with so many different
magnetic isotopes in the periodic table, something of chemical
significance would come to light? Rather like those early explorers, we
set out with the expectation that we would inevitably stumble on some
new land to call our own. Our trusted vessel was the "Pound
spectrometer" an oscillating detector9
that allowed one to search the largely empty radiofrequency spectrum in
the hope of finding the NMR response. Of course we knew roughly where
to look, for the values for these magnetic moments had already been
measured by zealous (but misguided) physicists. It was nevertheless a
real challenge to locate the tiny blip of an NMR signal in a vast sea of
noise, using a homemade spectrometer where the actual strength of the
magnetic field was only known very approximately. The Pound box
(sometimes known as a regenerative oscillator, autodyne detector, or
marginal oscillator) proved well suited to this task, making it easy to
sweep the radiofrequency by driving a tuning capacitor with a clock
motor and reduction gears. The regenerative feature of the Pound
spectrometer served to amplify weak NMR signals to the extent that no
further radiofrequency amplification was required. All that remained was
the straightforward task of relating the NMR frequency to a standard 5
MHz signal emitted by a radio transmitter located at Daventry.
Investigations were initiated on several of the "other" nuclear species - 7Li, 59Co, 69Ga, 71Ga, 113In, 115In, 203Th, and 205Th. Of these, the cobalt project proved to be the most rewarding. Norman Ramsey13
had just developed an important theory of chemical shifts. Even at the
time of the early molecular beam magnetic resonance experiments it was
understood that the magnetic field at the nucleus was slightly different
from the external applied field owing to the circulation of the atomic
electrons (the diamagnetic shielding). In molecules there is also a
paramagnetic term that reflects the fact that the electron distribution
around an atom that forms part of a molecule no longer has spherical
symmetry. Ramsey's calculation of this chemical shielding effect
involves a summation over all the excited electronic state
wavefunctions. He pointed out that the resulting chemical shift would be
particularly large (and temperature dependent) if there were a
low-lying electronic state, and suggested that this might occur in
cobalt (III) complexes. In this special case one term dominates all the
others, and the chemical shift should be inversely proportional
to the electronic energy gap Δ(sometimes called the ligand field
splitting) It is this particular electronic transition that gives rise
to the colour of these cobalt salts, so it can be measured in a standard
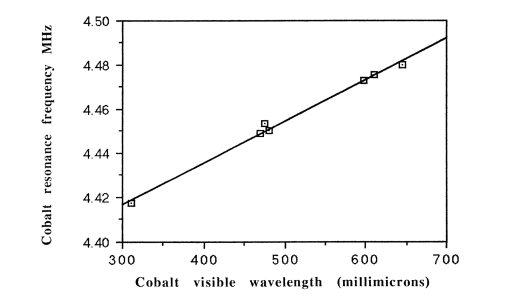
ultraviolet-visible spectrometer. We measured a series of symmetrical
cobalt complexes14
and found that the NMR frequencies (spanning the enormous range of
13,000 ppm) were directly proportional to the corresponding
ultraviolet-visible wavelengths, thus corroborating Norman Ramsey's theory. A physical chemist just loves a straight-line graph:
|
| |
 |
| |
|
To our chagrin, what seemed at the time to be a rewarding study on 59Co
NMR, was soon spectacularly upstaged by developments some 5,000 miles
away to the West. Scientists at Stanford University had just invented a
new form of spectroscopy that would change chemistry forever.
|
| |
4. High resolution NMR
In one of those fortunate coincidences that sometimes change the entire
direction of science, an Indian postdoctoral chemist Shrinivas
Dharmatti, happened to be working in the physics laboratory of Stanford
University. Having read about the chemical shifts observed by Warren
Proctor and others, he pointed out that almost any organic molecule
should show a proton spectrum consisting of many chemically shifted
lines, an idea that seems not to have occurred to the physicists at that
time for they had focussed on simple materials like water, and had not
paid much attention to seeking high resolving power. Using a very small
sample and painstakingly searching for a location in the magnet gap
where the field was most uniform, Martin Packard15
discovered the famous three-line spectrum of ethanol. Chemistry entered
a new golden age. Ask a freshman chemist, and he will happily write
down CH3CH2OH
for the structure of ethanol. Ask him how we know this to be the case,
and his justification becomes quite complicated, relying on a rich
history of interconnected reactions investigated by "wet chemistry",
including the oxidation of ethanol to acetaldehyde and acetic acid. In
contrast, here for the first time was direct evidence for three distinct
chemical sites, with the intensity ratio 3:2:1. Martin Packard (a
physicist) always asserts that he never believed the structural formulae
written by chemists until he saw for himself the three lines of
ethanol.
|
| |
5. Two key doctoral theses
Although the three-line ethanol spectrum is widely accepted as the
trigger for the NMR revolution that changed structural chemistry
forever, it was the doctoral theses of two young Stanford students, Jim
Arnold and Wes Anderson, that really demonstrated the incredibly broad
scope of this new form of spectroscopy. When their supervisor Felix
Bloch was appointed Director of the European Centre for Nuclear Research
(CERN) in Geneva, he took these research students with him, along with
their NMR equipment. There they wrote up their thesis work in two
classic papers19,20
that show for the first time the incredible potential of
high-resolution NMR. All aspiring NMR spectroscopists should read these
two seminal publications.
|
| |
5.1 James T. Arnold
An essential breakthrough was a remarkable permanent magnet designed and constructed by Jim Arnold19.
He set out to achieve a degree of uniformity of the magnetic field that
no one had ever dreamed was possible (approaching one part in 108).
Since the magnetic material (Alnico) had to be magnetized while the
magnet gap was closed, one pole of the magnet was mounted on roller
bearings, and once the magnet had been energized, the gap was opened
hydraulically (against a five-ton magnetic force of attraction) by means
of war surplus Boeing B-17 landing-gear struts. The initial charging
stage used a current of almost 400 amperes at a power level of 2
megawatts, and was carried out in the middle of the night to avoid any
serious disruption of the main Stanford electrical supply. The pole
caps, of very pure Armco ingot iron, were ground flat and parallel by an
outside contractor; everything else was done in the Stanford physics
laboratory. A permanent magnet is quite susceptible to field distortions
by nearby iron or steel objects such as tools, key rings, belt buckles
or pocket knives. In an amusing overreaction to this challenge, Jim
Arnold would strip to his underwear before operating the spectrometer.
Resolution was significantly improved by stirring the sample, an idea
that came to Felix Bloch21
one day while he was stirring his tea, surmising that this motion would
cause the nuclei to experience a magnetic field that was averaged over a
circular path. At first, stirring was achieved by means of a rotating
glass paddle inside the sample tube, but it was soon found that spinning
the cylindrical sample tube about its long axis was simpler and just as
effective. This soon became the accepted practice in all
high-resolution spectrometers. Great care was exercised in choosing the
materials of the radiofrequency probe since the presence of any
paramagnetic particles would be disastrous, and even diamagnetic
materials induce significant distortions of the field at the sample. All
this meticulous work culminated in a magnet with previously unheard-of
stability and uniformity, demonstrating for the very first time that an
NMR line could be recorded that was only 0.5 Hz wide. Even today this
would represent a perfectly acceptable resolving power.
For the very first time the rich complexity of a high-resolution NMR
spectrum became evident for all to see. In his new magnet Jim Arnold
showed that the three chemically shifted lines of ethanol carried
additional fine structure, attributed to a new kind of interaction
between adjacent nuclei. This "spin-spin coupling" indicated the number
of statistical combinations of "up" or "down" states of neighbouring
groups of protons. For example, the methyl resonance was split into a
1:2:1 pattern by the two protons in the adjacent methylene group. The
methylene resonance acquired a 1:3:3:1 pattern defined by the four
possible combinations of the spin orientations within the adjacent the
methyl group. Once spectroscopists had absorbed these new rules for
spin-spin multiplet structure, it came as a real disappointment to
discover further complicating effects attributable to "strong coupling".
These intensity distortions and new splittings could be calculated by
second-order perturbation theory19. In today's spectrometers, operating at far higher magnetic fields, strong coupling effects normally disappear.
Jim Arnold can also take credit for discovering the phenomenon of
chemical exchange. In a very pure sample of ethanol the rate of exchange
was slow enough that the coupling between CH2
and OH came into play - the hydroxyl resonance was a 1:2:1 triplet, and
the methylene response acquired an additional doubling. In contrast,
when very small concentrations of hydrogen ions were added, the exchange
rate was increased to the point that the OH resonance reverted to a
singlet and the corresponding splittings on the CH2
resonance disappeared. Fast exchange is said to "wash out" the
spin-spin splittings. Thus the first organic chemical chosen as an NMR
sample brought to light three new phenomena - proton spin-spin coupling,
strong coupling effects, and chemical exchange. The extraordinary
potential of NMR in chemistry was there for all to see.
|
| |
5.2 Weston A. Anderson
Wes Anderson's thesis explored the high-resolution spectra of several other simple organic compounds20.
In retrospect, his most important discovery concerned the first
proton-proton double resonance experiment. Although Robert Pound22 had earlier shown that the double resonance technique could be useful in NMR, Virginia Royden23 had decoupled the spin-spin interaction between 13C and 1H in an enriched sample of methyl iodide, and Arnold Bloom and Jim Shoolery24 had decoupled 19F from 31P, and 31P from 19F,
these were relatively straightforward heteronuclear experiments. Wes
Anderson's decoupling of one group of protons from another group of
protons presented a far more serious experimental challenge. His double
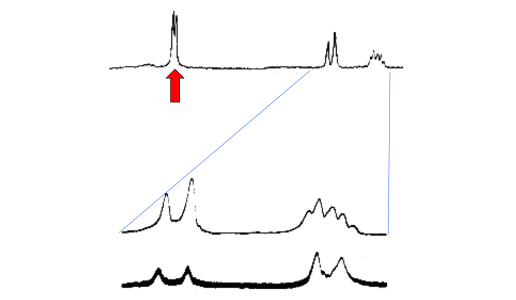
irradiation experiment20
on 2,3-dibromopropene (Figure 2) removed the overlying triplet
structure on the trans proton (bottom right) by selectively decoupling
the resonance of the CH2Br group (arrowed).
|
| |
 |
| |
In its simplest form homonuclear decoupling identifies groups that are
coupled by the spin-spin interaction; today we would say that these
chemical sites are correlated.
It is the philosophy behind these double resonance experiments
that has had the most important ramifications. At that period,
spectroscopy in general was straightforward - there was a direct
one-to-one relationship between sample and spectrum,
nothing could be changed in the spectrometer itself. Wes Anderson
demonstrated for the first time how proton NMR spectra could be manipulated
- altered by deliberate intervention of the spectroscopist to extract
further useful information. The manipulations soon became more
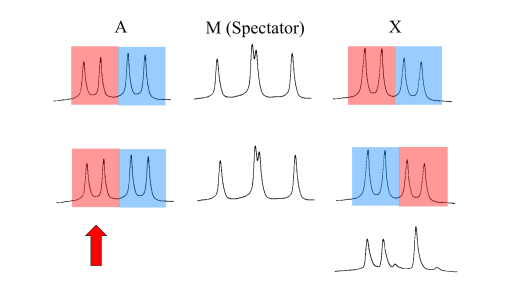
intricate. As one illustrative example, consider the three-spin "AMX"
proton spectrum25. of 2,3-dibromopropionic acid (Figure 3).
|
| |
| |
 |
| |
We know that the M nuclei consist of almost exactly equal
numbers of "up" and "down" spins. Suspend disbelief for a moment, and
assume that we can somehow physically separate the sample into two
flasks, one containing only the "up" M spins and the other the "down" M
spins. (We might usefully invoke James Clerk Maxwell's "demon" which
tests the second law of thermodynamics by identifying fast-moving gas
molecules and allowing them to pass through a trap door into a second
chamber, thereby warming the gas in that chamber.) In our proposed gedanken
NMR experiment, a sample taken from the flask with only "up" spins
could be examined in the NMR spectrometer. Disregard for the moment the M response; imagine M as merely a spectator. A sample taken from the first flask would now show the A and X
responses reduced to the "red" doublets, whereas a sample from the
second flask would show only the "blue" doublets. Our Maxwell demon has
succeeded in separating two subspectra. Now in making this assignment we
have tacitly assumed that the two coupling constants JAM and JMX
have like signs. If these signs are opposite, then the colour coding of
the upper trace should be changed to that shown in the lower trace. Of
course we cannot really call on the services of a Maxwell demon, but in
the real world the equivalent of this hypothetical experiment can indeed
be performed by double resonance. A selective irradiation experiment
can be set up to distinguish the upper spectrum of from the lower
spectrum. By suitably choosing the level of irradiation, it is feasible
to irradiate the "red" A doublet (red arrow) without appreciably affecting the "blue" A doublet, thus decoupling the "red" X doublet without affecting the "blue" X
doublet. The experimental double resonance response (Figure 3, bottom
right) corresponds unequivocally to the case of opposite signs.
Consequently JAM (a geminal coupling) is opposite in sign to JMX (a vicinal coupling). This result was of some significance at the time because theoretical calculations26 had predicted that geminal and vicinal couplings should all be positive.
This one example serves to demonstrate that manipulation of NMR spectra
by double resonance can reveal quite subtle properties of the molecule.
Extension of this technique by Forsén and Hoffman27
led to studies of slow chemical exchange, where irradiated spins were
physically transferred to a second chemical site by the exchange
process. A second powerful application, now widely employed for
structural studies, is the evaluation of proton-proton distances by the
nuclear Overhauser effect28. In a similar manner, broadband irradiation of protons is now used routinely to simplify 13C spectra and to obtain a roughly three-fold signal enhancement by the nuclear Overhauser effect29. Much later, in 1971, Jean Jeener30
showed that most double resonance experiments could be more efficiently
implemented by two-dimensional NMR, and this soon became the standard
"manipulation" procedure. Seldom is an NMR spectrum recorded today in
its pristine, unmodified, one-dimensional form. We could argue that Wes
Anderson's experiments provided the first impetus to the development of
the art of "spin choreography" that is fundamental to modern
multidimensional NMR. We have all become manipulators.
|
| |
6. Electromagnets
The high-resolution spectrum of ethanol highlighted the need for high
stability in any magnet designed for chemical applications of proton
magnetic resonance. There is no point in looking for fine detail on a
picture that is moving and shaking. Harry Weaver in the Stanford group
designed and built an electromagnet that operated at a proton frequency
of 30 MHz, chosen because the readily available war surplus radar
equipment employed an intermediate frequency amplifier (IF strip) at
this very frequency. The first step towards improving stability was to
regulate the current by means of eight parallel 304TL vacuum tubes.
(These later became an alarming feature of the early Varian
spectrometers; the electrodes become red-hot.) The next step was the
idea of "field/frequency" regulation, based on a proton "error signal"
from a second probe in the magnet gap. One such scheme is described in
an early patent by Russell Varian31 and the first practical implementation was demonstrated by Baker and Burd32. Another ingenious scheme was invented by Robert Pound15
who employed a pulse-modulated super-regenerative oscillator where the
phase of the radiofrequency pulses is triggered by the NMR signal
itself, so that the oscillator frequency automatically follows any
deviations of the magnetic field. Mention has already been made of the
"research student" drift compensation method employed in Rex Richards'
laboratory. In a similar spirit, Varian built a very effective
"super-stabilizer" that worked by sensing small field changes with a
pick-up coil feeding a sensitive galvanometer that shone a light beam
onto a pair of photocells. The field variations were compensated by a
correction current in coils attached to the pole faces. Wes Anderson16
devised a regulation scheme based on a "nuclear sideband oscillator"
that permitted the new Varian A60 spectrometer to use precalibrated
charts. It worked so well that Jim Shoolery tells the story that he was
amazed to be able to record an NMR spectrum twice, with the recorder pen
exactly retracing the first spectrum - proof that the regulation scheme
was very effective indeed. A final perfection was suggested by Hans
Primas17
who realized that an NMR signal from an internal reference (the twelve
equivalent protons of tetramethylsilane) would provide the best possible
regulation because it avoids the possibility of field changes between
two geometrically separated probes. This idea proved particularly useful
for performing frequency-sweep double resonance experiments18.
Stabilization via an internal reference signal soon became the accepted
practice; later the proton signal from tetramethylsilane was replaced
by a deuterium signal from a deuterated solvent, often CDCl3.
Spatial uniformity (homogeneity) of the magnetic field is the second
essential requirement. Not surprisingly, this represented an enormous
challenge to magnet technology. Expressed in terms of an astronomy
analogy, the resolution today achievable in NMR (approaching 1 part in
109)
would correspond to a telescope capable of resolving the images of two
cats sitting side by side on the surface of the moon. Before the work at
Stanford, no one had foreseen that this degree of field uniformity
could be achieved, or even felt that there would be any need for it.
Considerable thought was given to the design of pole caps, both with
respect to metallurgy and geometry, since these are critical in
determining the uniformity of the field. Electromagnets at that time had
to cycle the initial current according to a prescription that ensured a
slightly "dished" configuration of the magnetic field. A later
initiative was to shape the edges of the pole caps according to a
logarithmic function. Field distortions due to magnetic susceptibilities
of the copper wire used for the radiofrequency coil were eventually
corrected by devising a sandwich of two conducting materials with
opposite signs of their diamagnetic susceptibilities.
However it soon became clear that the most general solution to the
resolution problem was to accept that there would always be some
residual field inhomogeneities, but to compensate them empirically with a
set of correction coils. Based on calculations in terms of a spherical
harmonic expansion, Marcel Golay and Ed Jaynes independently designed
correction coils where the adjustments of individual gradients were
approximately independent. Golay33 made his first correction coils by embroidering them on calico; later printed circuits were used. Wes Anderson34
designed a set of coils that were generally adopted for all Varian
high-resolution magnets. There was much talk of orthogonality of these
"shim coils" although in practice the adjustment of the different field
gradients always involves some interaction, and considerable patience
and skill is still required to set up the most uniform magnetic field.
|
| |
7. Permanent magnets
As mentioned above, Jim Arnold opted for a permanent magnet, thus
avoiding many of the stability problems inherent in electromagnets. This
seemed like the preferred approach at that time, despite the fact that
the maximum achievable magnetic field was rather less than that of a
well-designed electromagnet. One early pioneer of high-resolution NMR,
Herbert Gutowsky35
adopted a permanent magnet for his home-built spectrometer.
Characteristically for a chemist, Gutowsky's turbines for spinning the
sample were carefully bored corks driven by an air jet.
At Oxford, Rex Richards36
also decided to assemble a high-resolution spectrometer based on a
prototype permanent magnet constructed by the Mullard Company for a sum
of £2,000; it operated at a proton frequency of 30 MHz. Since a
permanent magnet is quite sensitive to temperature, it was placed in an
insulated enclosure with thermostatic control. The detection of the NMR
signal was achieved by means of a commercial communications receiver.
The very first high-resolution spectrum was rather disappointing - it
seemed to be a band of pure noise - but closer examination showed some
narrow gaps where the noise had disappeared. The fact that the receiver
incorporated an automatic gain control had been overlooked, and once
this circuitry was disabled a beautiful high-resolution spectrum was
revealed.
|
| |
8. Commercial NMR Spectrometers
The full impact of NMR in chemistry had to await the advent of working
spectrometers that could be purchased from commercial companies, since
few chemists were prepared to make the big investment in time and effort
to build their own spectrometers from scratch. These instrument
companies included Varian Associates in California, Japan Electron
Optics Ltd. in Japan, Associated Electrical Industries in England,
Perkin Elmer Corporation in England, and Trüb Täuber (later Bruker
Spectrospin) in Switzerland. The complexity of these machines and the
limited market meant that this was not really a profitable business
venture. Varian NMR prospered by being part of a larger company that
made profitable microwave devices; indeed the Varian brothers (Russell
and Sigurd) had deliberately embarked on magnetic resonance in the full
knowledge that it would not be a viable commercial proposition. As
protection against competition, Varian Associates had acquired the
Bloch-Hansen patent that covered the NMR phenomenon itself, and Bloch's
spinning sample patent.
These early high-resolution spectrometers employed large electromagnets
and were not easy to operate or maintain. The best results required
careful regulation of the temperature of the water used to cool the
energizing coils, and often the magnet was housed in an insulating
enclosure to avoid the effects of changes in ambient air temperature.
Stability was greatly improved by the introduction of the
"superstabilizer" that monitored field changes and compensated them with
a current feedback scheme (see above). The definitive breakthrough came
with the introduction of the Varian A60 in 1961. This spectrometer was
designed specifically for the chemist who had no particular desire to
become an expert in radiofrequency electronics. It was a simplified,
slimmed-down version of the earlier Varian machines, with a much smaller
magnet, power supply and cooling system, all in a single enclosure. For
the first time, a minimalist design was adopted for the operating
controls, making this a truly user-friendly NMR spectrometer (some
critical controls were hidden behind a trapdoor to discourage
incorrigible knob-twiddlers). The console was dominated by a wide
flat-bed recorder that boasted a large paper chart precalibrated in ppm,
reproducibility being guaranteed by a regulation scheme based on Wes
Anderson's nuclear sideband oscillator. By defining a standard layout
for the NMR spectrum on a calibrated chart, this encouraged chemists to
acquire a "feel" for interpretation of spectra, and to adopt an
empirical approach to spectral assignment.
The A60 opened the way for a much wider acceptance of high-resolution
NMR among organic chemists. This was deliberately encouraged by the
Varian personnel, who toured the chemistry departments of universities
and industries to explain the basic principles of this alien new
technology, and to demonstrate how beautifully it could solve structural
problems. They were led by a great enthusiast, Jim Shoolery, one of the
first chemists to fully appreciate the enormous potential of the
technique. Every effort was made to keep the talks pictorial and
non-mathematical, as a profound knowledge of the fundamental physics was
not considered essential for the acquisition of NMR spectra. This
educational initiative was reinforced by the introduction of a series of
NMR "workshops" where owners and prospective owners could acquire
"hands on" experience of the new machines. Although limited to proton
studies, this can be regarded as the golden age of high resolution NMR.
Almost any organic compound taken down from the shelf would yield an
interesting proton spectrum.
|
| |
9. Sensitivity
Even during the euphoria of the early 1960's some scientists began to
ponder the question of poor NMR sensitivity, limited by the low
Boltzmann factor of nuclear spin populations at room temperature. The
first attack on this problem came from Klein and Barton37
who converted a digital pulse-height analyser into a device that added
together successive NMR traces, thereby increasing the signal-to-noise
ratio by the square root of the number of accumulations. At an American
Chemical Society "Meeting in Miniature" in December 1962, the audience
was amazed by Melvin Klein's practical demonstration of an NMR trace
that improved in quality before our very eyes. This seemed like magic.
About the same time Jardetsky et al38
employed a commercially available Mnemotron "computer of average
transients" to enhance the NMR signals of some biochemical samples. Very
quickly the time averaging method was generally adopted. The only
drawback was the law of diminishing returns imposed by the square root
dependence; high signal enhancement factors inevitably meant
unacceptably long time-averaging durations. The practical limit appeared
to be signal accumulation over an entire weekend, provided that the
spectrometer was not required for more important work.
|
| |
9.1 Multichannel excitation
It was Wes Anderson who realized that the conventional field sweep (or
frequency sweep) procedure was inherently inefficient with regard to
sensitivity. At any given instant, the spectrometer receiver detects the
intensity at only one point on a particular resonance line in a
spectrum of many lines, indeed at other instants it wastes time
gathering only baseline noise. Wes suggested that a multichannel
irradiation device that excited all frequency regions simultaneously
would be far more efficient. With N independent excitation channels the signal-to-noise ratio should improve as √N,
up to the approximate limit of one channel per line width. Wes loves
mechanical gadgets, so he set out to design a device to generate a set
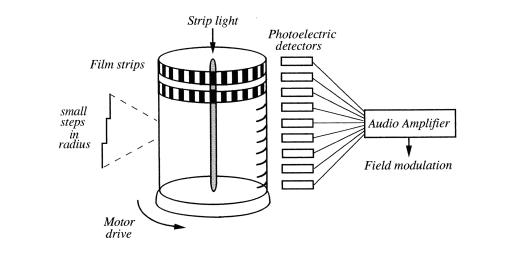
of N regularly spaced audio frequencies. A rotating hollow Lucite
cylinder had a strip light running down the central axis (Figure 4).
|
| |
 |
| |
The outer circumference was machined horizontally in 20 vertical steps
with linearly increasing radii. Strips of photographic film with
alternating dark and light bands were glued around the different levels
so that as the cylinder rotated, the light that fell on a vertical array
of photoelectron detectors was chopped at a set of gradually
incremented frequencies. These were then used to generate a regular
"comb" of modulation sidebands that covered the entire proton NMR
spectrum. The same cylinder could be used in reverse to demodulate the
received NMR signal. The device became known as the "Prayer Wheel",
being reminiscent of those used by Buddhist monks to enhance their
prayers by constant repetition.
The prayer wheel was never used in anger because a more promising
method of broadband excitation emerged, and Wes reluctantly allowed his
brainchild to be taken away and installed in the Smithsonian Institution
in Washington. This may stand as a lesson to all, that certain seminal
ideas can turn out to be more important than their actual practical
implementation. Much later it became possible to vindicate the concept
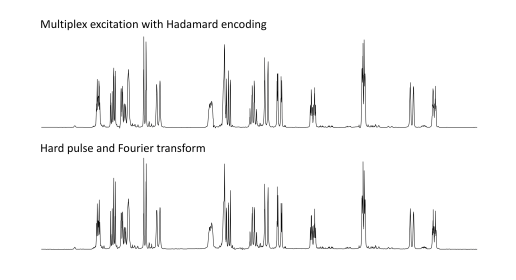
of multichannel excitation by using modern waveform generator technology
to excite part of the 500 MHz proton NMR spectrum of strychnine (Figure
5) with 2048 separate irradiation frequencies spaced 1Hz apart, using
Hadamard encoding to differentiate the resulting 2048 responses39.
There is no perceptible difference between this multiplex spectrum and
that recorded by the conventional Fourier transform method.
|
| |
 |
| |
9.2 Fourier transform NMR
The approach that superseded the prayer wheel was based on an idea for
broadband excitation of the entire NMR spectrum by an intense
radiofrequency pulse, followed by Fourier transformation of the
resulting transient free induction decay. This concept was first
formulated by Russell Varian in a patent filed on 29 August 1956. This
revolutionary patent even contained suggestions for an analogue Fourier
transformation scheme based on repetitive reading of the transient free
induction signal stored on a loop of magnetic tape, picking out the
individual frequency components by means of a variable-frequency lock-in
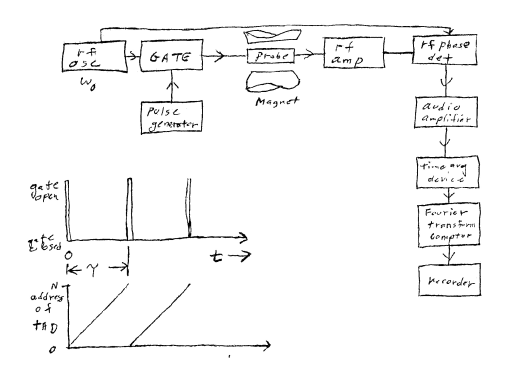
detector. In his laboratory notebook (dated 3 June 1964) Wes Anderson
sets out a practical block diagram (Figure 6) for pulse excitation of
all the different NMR frequencies in a proton spectrum, and a scheme for
sorting the different frequency responses by Fourier transformation.
|
| |
 |
| |
The method relies implicitly on two important features - time averaging
of many successive free induction decays, and the availability of a
suitable computer for Fourier transformation.
Practical implementation of the concept was greatly hampered by the
complications involved in digitising the transient NMR signal, and the
difficulty in finding a computer to perform the Fourier transforms.
Small laboratory computers had yet to be invented. One very cumbersome
approach - the spectrum analyser, repetitively examines the time-domain
signal and slowly picks out the component frequencies one by one. It is
interesting to note that the first digital computer programs for Fourier
transformation employed an analogous stepwise examination of the
spectral frequencies, entailing a delay of 20 minutes or so for a
typical transformation. Completion of the transform in a more reasonable
time (seconds) had to await the Cooley-Tukey fast Fourier transform
algorithm40.
At Varian at that time it happened that Richard Ernst had been working
on methods to improve the sensitivity of the conventional NMR sweep
protocol by optimizing the various operating parameters, so he was
naturally well primed to attack the proposed new Fourier transform
scheme. It is greatly to the credit of Ernst and Anderson41
that they persevered with this daunting project even though it appeared
rather impractical at the time. Digitization and temporary storage of
the accumulated transient NMR signal involved a tedious conversion,
first to paper tape and then to IBM punched cards - a whole box of cards
was used to carry a single free induction decay to a central computer
located at the far end of Palo Alto. The large IBM 7090 computer at the
local Service Bureau relegated such "trivial" calculations to overnight
runs. Typically, the first spectrum would need readjustment of the phase
(and the phase gradient) to give pure absorption-mode signals,
requiring another round trip to the service bureau. Not many chemists
were prepared to wait a matter of days for a properly phased NMR
spectrum. Nevertheless the final results were impressive - an order of
magnitude improvement in proton sensitivity compared with the
conventional sweep method executed in the same total time. It is a sad
reflection on our peer-review system that referees twice
contrived to find reasons to reject Ernst and Anderson's manuscript
submitted to a respected physical chemistry journal; the story
eventually appeared in press in 1966. NMR spectroscopy was changed
forever.
The most significant outcome of this new Fourier transform spectroscopy
was a gift to the chemists that they had always desired, but had been
afraid to request - 13C
spectra from samples of natural isotopic abundance (1.08%). Carbon
defines the basic framework of any organic molecule. Because 13C
spectra are simpler and the information is more widely dispersed than
in proton spectra, they offer a more reliable fingerprint for
identification and structural determination. The Fourier transform made
this possible, aided by an important spectral enhancement technique -
broadband irradiation of the protons, which greatly simplifies 13C spectra by removing the 13C-H
fine structure and inducing a roughly threefold signal enhancement
through the nuclear Overhauser effect. The first flowering of NMR in
chemistry was now virtually complete.
|
| |
Acknowledgements
The Encyclopedia of Magnetic Resonance, Volume 1: Historical Perspectives
(John Wiley & Sons, 1996; Editors in Chief: David M. Grant and
Robin K. Harris) proved very useful for checking chronology, references,
and some half-remembered facts about the early days of NMR in
chemistry. I thank the NMR pioneers (Rex Richards, Anatole Abragam,
Ionel Solomon, Bob Pound, John Pople, David Whiffen, Warren Proctor,
Martin Packard, Jim Arnold, Wes Anderson, Jim Shoolery, Richard Ernst,
Jean Jeener) all of whom I was fortunate to know personally, and I ask
for their forbearance if my memory of some details has not been as
accurate as they might wish.
|
| |
References
1. E. M. Purcell, H. C. Torrey and R. V. Pound, Phys. Rev. 69, 37 (1946).
2. F. Bloch, W. W. Hansen and M. E. Packard, Phys. Rev. 69, 127 (1946).
3. W. G. Proctor and F. C. Yu, Phys. Rev. 77, 717 (1950).
4. W. C. Dickinson, Phys. Rev. 77, 736 (1950).
5. G. Lindström, Phys. Rev. 78, 817 (1950).
6. H. A. Thomas, Phys. Rev. 80, 901 (1950).
7. G. E. Pake, J. Chem. Phys. 16, 327 (1948).
8. H. S. Gutowsky, G. B. Kistiakowsky, G. E. Pake and E. M. Purcell, J. Chem. Phys. 17, 972 (1948).
9. R. V. Pound, Phys. Rev. 72, 527 (1947).
10. R. Freeman and R. E. Richards, Trans. Faraday Soc. 52, 802 (1956).
11. J. S. Waugh, L. M. Huber and U. Haeberlen, Phys. Rev. Lett. 20, 180 (1968).
12. P. Mansfield, Phys. Lett. 32A, 485 (1970).
13. N. F. Ramsay, Phys. Rev. 78, 699 (1950).
14. R. Freeman, G. R. Murray and R. E. Richards, Proc. Roy. Soc.(London) A. 242, 455 (1957).
15. R. V. Pound and R. Freeman, Rev. Sci. Instr. 31, 96 (1960).
16. W. A. Anderson, Rev. Sci. Instr. 33, 1160 (1962).
17. H. Primas, Fifth European Congress on Molecular Spectroscopy, Amsterdam (1961).
18. W. A. Anderson and R. Freeman, J. Chem. Phys. 37, 85 (1962).
19. J. T. Arnold, Phys. Rev. 102, 136 (1956).
20. W. A. Anderson, Phys. Rev. 102, 151 (1956).
21. F. Bloch, Phys. Rev. 94, 496 (1954).
22. R. V. Pound, Phys. Rev. 79, 685 (1950).
23. V. Royden, Phys. Rev. 96, 543 (1954).
24. A. L. Bloom and J. N. Shoolery, Phys. Rev. 97, 1261 (1955).
25. R. Freeman and D. H. Whiffen, Mol. Phys. 4, 321 (1961).
26. M. Karplus, J. Am. Chem. Soc. 84, 2458 (1962).
27. S. Forsén and R. A. Hoffman, Acta Chem. Scand. 17, 1787 (1963).
28. F. A. L. Anet and A. J. R. Bourn, J. Am. Chem. Soc. 87, 5250 (1965).
29. K. F. Kuhlmann and D. M. Grant, J. Am. Chem. Soc. 90, 7355 (1968).
30. J. Jeener, Ampere International Summer School, Basko Polje, Yugoslavia (1971).
31. R. H. Varian, U.S. Patent 3,109,732, Filed 29 August 1956.
32. E. B. Baker and L. W. Burd, Rev. Sci. Instr. 28, 313 (1957).
33. M. J. E. Golay, Rev. Sci. Instr. 29, 313 (1958).
34. W. A. Anderson, Rev. Sci. Instr. 32, 241 (1961).
35. H. S. Gutowsky C. J. Hoffmann and R. E. McClure, Phys. Rev. 81, 305 (1951).
36. J. B. Leane, R. E. Richards and T. P. Schaefer, J. Sci. Instr. 36, 230 (1959).
37. M. P. Klein and G. W. Barton, Rev. Sci. Instr. 34, 754 (1963).
38. O. Jardetsky, N. G. Wade, and J. J. Fischer, Nature (London) 197, 183 (1963).
39. E. Kupce and R. Freeman, J. Magn. Reson. 162, 158 (2003).
40. J. W. Cooley and J. W. Tukey, Math. Comput. 19, 297 (1965).
41. R. R. Ernst and W. A. Anderson, Rev. Sci. Instr. 37, 93 (1966).
|
| |
|
|
 |
| Ray Freeman |
| |
|